SEPARATION OF PHOSPHOLIPIDS
BY TLC
A particularly frequent approach is to obtain information on the various phospholipid components of the lipid extract under investigation. This can be achieved easily and efficiently by subjecting a sample to thin-layer chromatography (TLC).
There is a huge number of TLC techniques described for that purpose, some of them using one-dimensional migration others using two successive migrations in two orthogonal directions (two-dimensional).
We describe below some techniques that have proven easy and reliable in our hands but others could be found in specialized books and scientific journals.
The proposed simple one-dimensional TLC procedure is routinely used in our laboratory and may be considered as efficient as common two-dimensional techniques (Leray C et al., J Chromatogr 1987, 420, 411). It has the peculiarity to allow a good separation of phosphatidylinositol and phosphatidylserine owing to the impregnation of TLC plates with boric acid.
Other one-dimensional techniques are used for the separation of less frequent phospholipid forms (polyphosphoinositides, mono-and dimethyl phosphatidylethanolamine …) or for quantitative estimation by densitometry.
For some specialized purposes, a two-dimensional procedure is also described.
One-dimensional TLC
Separation of phosphoinositides
Separation of methylated phosphatidylethanolamines
![]()
One-dimensional TLC
The TLC described below allows an efficient separation of all the main phospholipid classes for their quantification by phosphorus assay or fatty acid analysis (Leray C et al., J Chromatogr 1987, 420, 411).
Apparatus:
Whatmann LK5 thin-layer plates with a kieselguhr concentration zone, glass chromatographic tanks for pre-washing, preservation and solvent migration. Calibrated syringes (10-25 µl)
Reagents:
Solution of 2.3% (w/v) boric acid in ethanol.
Chloroform, ethanol, triethylamine (keep at 4°C), primuline solution for spray (5 mg in 100 ml of acetone/water, 80/20, v/v), methanol.
Procedure:
TLC plates are pre-washed by migration up to 1 cm from the top in a clean tank containing chloroform/methanol (1/1, v/v). Plates are air-dried in a fume hood and placed in the preservation tank until used. This step is required to remove any material interfering with further quantitative and qualitative analyses.
Before use, plates are thoroughly wetted (we use a plastic bottle with a plunger tube) with boric acid solution, drained 5 minutes in a fume hood and dried for 15 min at 100°C in an oven.
Lipid samples (10 up to 100 µl chloroform solution containing 20-200µg phospholipids) are rapidly deposited on plates as 1 or 2 cm parallel streaks in the concentration zone.
Plates are rapidly placed in the chromatography tank containing the following solvent:
chloroform/ethanol/water/triethylamine (30/35/7/35, v/v). The migration time is close to 2 hours. Plates are dried in a fume hood (2-5 min max) and sprayed with primuline solution. After viewing under UV light, fluorescent spots are scraped into glass tubes.
Phosphorus contents are determined after mineralization with perchloric acid and fatty acids are analyzed as previously described.
If required for further studies, phospholipids are extracted from the scraped silica gel by two elutions with 5 ml of chloroform/methanol/water (5/5/1, v/v) and a further addition of 5 ml water with mixing to the combined supernatants. The lower phase, collected after centrifugation, is evaporated. Lipids are dissolved in a small volume of chloroform/methanol (2/1, v/v). An antioxidant (BHT) is added (final concentration: 1 mg/ml) if lipids are stored for more than one day even at -20°C.
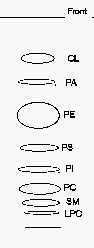
TLC separation of phospholipid classes
CL: cardiolipin, LPC: lysophosphatidylcholine, PA: phosphatidic acid, PC: phosphatidylcholine, PE: phosphatidylethanolamine, PI: phosphatidylinositol, PS: phosphatidylserine, SM: sphingomyelin.
Comments: to get an optimal detection under UV lamp, the plates should smell moderately triethylamine before spraying since this chemical quenches the induced fluorescence.
If a better separation of the phospholipids with the lowest Rf is needed, a second run in the same solvent following a rapid drying is advisable.
As the solvent mixture is poorly volatile, the solution can be used for several runs for at least two days.
When phosphatidylglycerol (PG) is present in the lipid sample, it can be detected between PE and PS. When an acidic treatment is processed before the TLC step, a spot of lysoPE produced by plasmalogens may be found between the spots of PS and PI.
A modification of our procedure was proposed using plates previously wetted with 1.8% boric acid instead of 2.3% as written above (Vaden DL et al., Anal Biochem 2005, 338, 162). With that small modification, the authors obtained a better separation of PE from PG, and of PA from monolysocardiolipin.
![]()
Quantification of phospholipid classes by scanning densitometry.
This approach involving a photodensitometry of charred lipids directly on TLC or better on HPTLC plates is convenient for large numbers of samples or for samples containing low amounts of phospholipids. Several manufacturers (Camag, Shimadzu) produced reliable computer-controlled densitometers but pictures captured with a digital camera may be also proposed in combination with adapted softwares. In the absence of a scanning densitometer, the quantitative determination of phospholipid distribution may be realized with a liquid scintillation counter commonly used for radioactivity measurements (Shand JH et al., Anal Biochem 1980, 101, 427).
Procedure:
HPTLC plates (10×20 cm or 10×10 cm) from Merck are pre-washed by migration in chloroform/methanol (1/1, v/v). Apply no more than 30 µg phospholipids as a 8 to 10 mm streak at 1.5 cm from the bottom of the plate. Place in a small TLC tank with two sides lined with filter paper and containing a small volume of the mixture described by Heape et al. (J Chromatogr 1985, 322, 391): methyl acetate/n-propanol/chloroform/methanol/0.25% KCl in water (25/25/28/10/7).
The migration time is about 45 min up to 1 cm from the top.
After drying, the TLC plates are treated by various stains but two are commonly used:
1. Dittmer solution for phosphorus determination
2. Charring after spraying with a cupric solution.
Plates are largely sprayed until translucent with a 10 % (w/v) cupric sulfate solution in 8% (w/v) orthophosphoric acid (Baron et al., J Liquid Chromatogr 1984, 7, 2793).
Plates are heated in an oven at 180°C for 10-15 min. This time should be adapted according to the TLC plate brand in order to minimize the background staining. Some plate brands contain organic binder which can darken at elevated temperatures.
We have selected this copper reagent since it was proved to be the most independent of the number of double bonds in the lipid samples. As plates vary in response, standards must be included in each run. Phospholipid classes give roughly the same response except for sphingomyelin which gives higher responses. No more than 4-5 µg phospholipids per spot can be scanned but very low amount are detectable.
A similar procedure based on another solvent mixture has been described for the analysis of red cell membrane phospholipids (Weerheim AM et al., Anal Biochem 2002, 302, 191).
![]()
Separation of phosphoinositides.
To study the polyphosphoinositides labelled with 32P either in cultivated cells or under the action of a preparation of phosphatidylinositol-3-kinase, we use a very efficient TLC procedure with the mixture : chloroform/methanol/water/conc. NH4 (45/35/8.5/1.5, v/v) as solvent system. Better results are obtained when the chromatography tank is previously placed at 4-6°C. The running time is evidently longer than at room temperature. The temperature of the cold room must not be too low since two phases are formed in the tank.
To study the highly phosphorylated phosphoinositides, it is recommended to use another TLC system which separates efficiently the triphosphoinositides from the radioactivity remaining near the origin of the run and any radioactive ATP and phosphate carried over with the organic phase (Van Dongen CJ et al., Anal Biochem 1985, 144, 104).
Procedure:
The TLC plates (silica gel 60 Merck) are pre-coated with a mixture of methanol/water (2/3) containing 1% potassium oxalate (w/v) and 1 mM EGTA (the solution is poured on the plate using a pipette). The plates are dessicated at 100°C for 20-30 min before use.
Plates are run in the solvent system : chloroform/acetone/methanol/acetic acid/water (46/17/15/14/8, v/v). The solvent is allowed to migrate nearly to the top of a 20 cm TLC plate. The plates are then thoroughly dried in a well ventilated hood (to remove acetic acid) and the lipid spots detected after a primuline spray. The observed Rf of the phosphoinositides are: PI 0.5; PIP 0.33; PIP2 0.25 and PIP3 about 0.2. Standards are commercially available (see links: Echelon Research Laboratories Inc). These phosphoinositides are better localized by autoradiography after labeling cells with radioactive phosphate (Figure below).

The phospholipid spots are carefully scraped in a glass screw-capped tube and either extracted or deacylated according to the running study (fatty acid analysis, radioactivity measurement or analysis of polyphosphoinositides).
When the phospholipids have to be recovered from silica gel, the scraped spots are extracted 2 times with 2 ml of chloroform/methanol/conc. HCl (20/40/1, v/v/v) with the help of sonication and vortexing. Wait about 1 h between the two extractions. To the supernatant (4 ml), add 1.3 ml chloroform and 2.4 ml water. After vortexing and centrifugation, the lower phase is collected and concentrated.
The study of the isomeric forms of phosphoinositides is done after a mild deacylation of phospholipids and by HPLC separation of the derived glycerophosphoinositol phosphates.
Alternatively, a protocol using TLC to separate phosphoinositide classes according to their head groups and quantitative GC of fatty acid methyl esters from lipids isolated has been proposed (Konig S et al., Anal Biochem 2008, 378, 197). Phosphatidylinositol monophosphates and bisphosphates were separated using two different solvent systems. Sample lipids were scraped and analyzed for their fatty acid contents.
![]()
Separation of methylated phosphatidylethanolamines
The metabolic transformation of phosphatidylethanolamine into phosphatidylcholine through three methylation steps is of prime importance in the nervous system.
To separate the two intermediates from the substrate and the final reaction product, we propose a simple mono-dimensional system using the mixture: chloroform/propionic acid/n-propanol/water (2/3/2/1, v/v) as solvent system. This system requires a running time of about 4 h.
Before viewing the phospholipid spots after primuline spray, it is necessary to dry thoroughly the plates 15 min in a fume hood and under vacuum for 30 min before primuline spray.
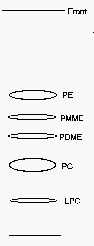
PE: phosphatidylethanolamine, PMME: phosphatidyl monomethylethanolamine, PDME: phosphatidyl dimethylethanolamine, PC: phosphatidylcholine, LPC: lysophosphatidylcholine.
![]()
More complex lipid components can be separated on a single TLC plate with a two-dimensional system. Several systems have been devised with a variety of solvent combinations. The most efficient system involved contrasting solvents, i.e. a neutral or basic solvent in the first direction followed by an acidic solvent in the second direction.
The principal disadvantage of the technique is that the precise orientation of the separated components on the plate may vary from one run to another and no standard sample can be used to appreciate the variations. The double development is also time-consuming. In some special occasions this two-dimensional system is of special interest. We give below two examples of two-dimensional TLC system we used for the separation of methylated phosphatidylethanolamine and for trinitrobenzene derivatives of phosphatidylserine and phosphatidylethanolamine.
![]()
Method 1
Separation of methylated phosphatidylethanolamines
Apparatus
Silica gel G60 plates (Merck) 20×20 cm
Plastic tank to maintain silica gel plates under vacuum (motor pump)
Reagents
– solvent for the first dimension: chloroform/methanol/conc ammonia (65/35/4, v/v)
– solvent for the second dimension: butanol/acetic acid/water (60/20/20, v/v)
Procedure
Since this method is mainly used to separate radioactively labelled components, small amounts of mono- and dimethyl-phosphatidylethanolamine are added to the samples. No more than 1 mg phospholipids are applied as a small spot in a corner of the plate at 1.5 cm of each side. After the first dimension, the plate is dried 5 min in a fume hood and 15 min in the vacuum tank. After the second dimension, dry the plates for 330 min under vacuum before the primuline spray.
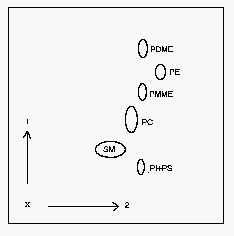
1: first dimension, 2: second dimension, PI: phosphatidylinositol, PS: phosphatidylserine, SM: sphingomyelin, PC: phosphatidylcholine, PE: phosphatidylethanolamine, PDME: dimethyl phosphatidylethanolamine, PMME: monomethyl phosphatidylethanolamine.
![]()
Method 2
Separation of phospholipids and their trinitrobenzene derivatives
Apparatus
As described before in method 1
Reagents
– Solvent for the first dimension: methyl acetate/n-propanol/chloroform/methanol/0.25% KCl (w/v) in water (25/25/28/10/7, v/v)
– solvent for the second dimension: chloroform/methanol/acetic acid/water (90/40/12/2, v/v)
Procedure
As describe before for method 1
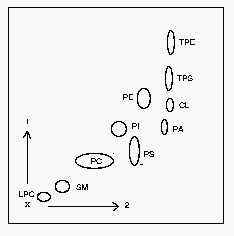
1: first dimension, 2: second dimension. CL: cardiolipin, LPC: lysophosphatidylcholine, PA: phosphatidic acid, PC: phosphatidylcholine, PE: phosphatidylethanolamine, PI: phosphatidylinositol, PS: phosphatidylserine, SM: sphingomyelin, TPE: trinitrobenzene derivative of PE, TPS: trinitrobenzene derivative of PS.
DISPERSIVE LIQUID-LIQUID MICROEXTRACTION
Lire la suiteDevenez membre et participez au développement de la Lipidomique au XXIème siècle.
S'inscrire