VITAMIN K ANALYSIS
Vitamin K is present at extreme low concentrations relatively to the lipid compounds present in plasma or animal tissues and, thus, requires for its precise determination a reliable extraction process and a combination of several chromatographic systems.
Numerous technical reports emerged in the 1980s based on various detection systems (ultraviolet, electrochemical, fluorimetry). However, it is now recognized that the methodology based on reduction of the vitamin K coupled with the fluorimetric detection of its fluorescent hydroquinone derivative is the most selective and sensitive application.
This new procedure is based on a methodology originally described by Haroon Y et al. (Clin Chem 1986, 32, 1925). A sensitive procedure using an on-line photoreactor and peryoxalate chemiluminescence detection has also been described (Ahmed S et al., Anal Chim Acta 2007, 591, 148).
The analytical system utilized a dry chemical reactor (small column) containing powdered zinc between the column and the fluorescence detector. This analytical system converts vitamin K compounds into their fluorescent hydroquinones by reduction over zinc in the presence of zinc ions in acidic medium.
As several interfering substances are present in biological materials, crude lipid extracts cannot be used for direct HPLC analysis. Thus, it is imperative to purify samples and concentrate the vitamin K amounts before the analytical step.
For liver samples, we have adopted a preliminary HPLC step on a silica column with collection of a vitamin K-containing fraction followed by a HPLC separation on a reversed-phase column. It is recommended to use dedicated HPLC system for each stage of the assay. Other sample extraction methods were proposed for HPLC determination of phylloquinone and menaquinones including solvant partition, thin-layer chromatography and solid-phase extraction. Some of these techniques will be examined later.
Determination of vitamin K1 and VK oxide
Equipment and reagents
Normal phase column: Lichrospher Si-60, 5 µm, 250×4 mm, Merck
Reversed-phase column: Lichrospher 100RP-18, 5 µm, 250×4 mm, Merck
Reducer column: a 2 x 0.4 cm column dry-packed using vibration with high-purity zinc metal (200 mesh). This device may be found at INTERCHIM (Ref. CH18541-ZN)
Silica SPE cartridge
Hexane, ter-butyl methyl ether, diethyl ether, ethanol, methanol, tetrahydrofuran (THF),
NaClO4
Tissue extraction
Liver tissue: a sample (about 1 g) is finely powdered in a mortar cooled with liquid nitrogen. The powder is rapidly transferred in a weighed amber tube containing 3 ml ethanol and 2 ml water, 8 ml of hexane/ether (v/v) are then added and the mixture is vortexed for 10 min. After a short centrifugation, the upper phase is removed, evaporated under a stream of nitrogen and the lipid extract is dissolved in 400µl of hexane.
Plasma: 500 µl are mixed with 1 ml ethanol. The mixture is mixed for 15 min after addition of 5 ml of hexane. The hexane layer is removed after a short centrifugation, evaporated under nitrogen and dissolved in 100 µl of hexane.
Lipid extracts from plasma can be processed through a simple purification process involving SPE cartridges. The hexane extract is filtered on a 500 mg silica cartridge previously washed with 5 ml of hexane. After filtration of the extract, the column is washed with 10 ml of hexane, the vitamin K fraction being eluted with 5 ml of hexane/ether (97/3, v/v), the last 4 ml of the eluates are evaporated and analyzed by HPLC as in the second analytical step.
Others (Davidson K et al., Meth. Enzymol. 1997, 282, 408) claim that a purification on a C18 SPE cartridge , as described for food extracts, is efficient before the analysis of plasma or serum extracts. The final lipid residue must be first dissolved in 25 µl dichloromethane, immediatly followed by 175 µl methanol and then analyzed on a reversed-phase column.
Food: Crude lipid extracts of meat, foods, milk or vegetables cannot be used for direct HPLC analysis of vitamin K compounds since they cannot be isolated from triglycerides using saponification owing to their instability under alkaline conditions. Lipase treatment was proposed (Barnett SA et al., Anal Chem 1980, 52, 610; Indyk HE et al., J AOAC Int 1995, 78, 719) but solid-phase extraction is a more effective and more simple purification step procedure.
Procedure: One gram of food is finely powdered in a mortar cooled with liquid nitrogen. For vegetables and fruits (if high water content), grounding is made in the presence of 10 times the weight of anhydrous sodium sulfate. After transfer in a 50 ml polypropylene tube, 15 ml of 2-propanol/hexane (3/2, v/v) are added, followed by 4 ml water (32 ml when sodium sulfate is used). The mixture is sonicated for 30 sec and vortexed for 10 min. After centrifugation, the upper phase is collected and evaporated to dryness under a stream of nitrogen. Residues are dissolved in 2 ml of hexane for SPE. If vegetable oil is analyzed, a 0.1 ml aliquot is directly dissolved in 4 ml hexane.
The lipid extract is applied to a 3 ml (500 ml) SPE silica column (a 6 ml column is used for meats, fats and oils). After preconditioning the column with 8 ml of hexane/diethyl ether (97/3, v/v) and 8 ml pure hexane, the extract is filtered through the column, followed by a wash with 8 ml of hexane. The vitamin K-rich fraction is eluted with 8 ml of hexane/ether (97/3, v/v).
For lipid-rich samples, an additional purification step on a 6 ml (1 g) C18 SPE column is required. The residue of the silica SPE column is slowly dissolved in 0.2 ml of 2-propanol while heating slightly. This extract is applied to a C18 SPE column preconditioned by washing with 10 ml of methanol/dichloromethane (8/2, v/v), followed by 6 ml of methanol and 6 ml of water. After filtration of the extract, the column is washed with 6 ml of methanol/water (95/5, v/v), followed by 6 ml of acetonitrile. The vitamin K-rich fraction is eluted with 10 ml of methanol/dichloromethane (8/2, v/v).
The eluants are evaporated to dryness and dissolved in the mobile phase of the reversed-phase chromatography.
Bones: Samples (about 1 g) are finely powdered in liquid nitrogen with a special freeze mill. Powder is washed 3 times with water (40 ml) and lyophilized. An aliquot is extracted with 20 ml chloroform/methanol (3/1, v/v) by prolonged vortexing. The extracted powder is dessicated and demineralized for a night at 4°C in 10 ml of 0.5 M EDTA. The water suspension is extracted with 40 ml of chloroform/methanol mixture. The extracts are evaporated under vacuum on a rotary evaporator (protected from light). The lipid extract is hydrolyzed using lipase from Candida cylindricea (Type VII from Sigma) (5 ml of 0.8 M phosphate buffer pH 8 containing 0.5 g lipase for 1 h at 37°C. The incubation medium is extracted with 30 ml hexane after addition of 10 ml ethanol. After centrifugation, the upper phase is evaporated, re-suspended in 5 ml hexane and purified on silica SPE column as for food extracts.
For the separation of menaquinones contained in bone extracts, see the works of Hodges SJ (J Bone Mineral Res 1993, 8, 1005) on human bone or of Tamaoka J (Meth Enzymol 1986, 123, 251) on bacteria.
Purification (semi-preparative chromatography)
Lipid extracts from liver (100 µl) are injected on a silica column (Lichrospher Si-60) eluted with the mixture hexane/ter-butyl methyl ether (99.3/0.7, v/v) at 1.5 ml/min. The fraction containing vitamin K1 and vitamin K1 oxide (between about 12 and 18 mn) is collected in an amber vial. This fraction is localized by UV spectrophotometry (246 nm) after supplementation of a similar sample with 1-2 µg vitamin K1 and its oxide. The collected solvent is evaporated under nitrogen and the extract is dissolved with 200-500 µl methanol.
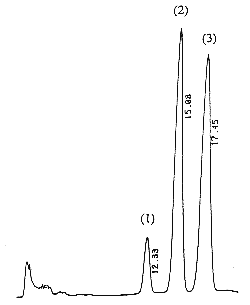
A chromatogram of cis (peak 1), trans (peak 2) vitamin K1 and vitamin K1 oxide (peak 3) as standards is given below (optimal detection for the epoxide: 259 nm).
The column is then washed by pumping THF for 10 min (2 ml/min) followed by the chromatographic eluant for 15 min (2 ml/min) in open circuit and for a minimum of 10 min (1.5 ml/min) in closed circuit (equilibration step).
Analytical chromatography
The fraction collected in the previous step is injected (50 µl) on a reversed-phase column with methanol/water (99.7/0.3, v/v) containing 20 mM NaClO4 as the mobile phase (flow rate: 1 ml/min). After reduction in the zinc column, the fluorescent molecules are detected with a fluorometer (excitation: 320 nm, emission: 430 nm).
The retention times of vitamin K1 oxide and vitamin K1 are about 8 and 14 min, respectively.
To help the localization of the two compounds, it is recommended to collect several 2 ml fractions in the expected elution zone during the normal phase chromatography and then to analyze these fractions by reversed-phase chromatography. An example of this purification test is given below (6 fractions were collected from the appearance of vitamin K1 after a 12 min elution on a normal phase column).
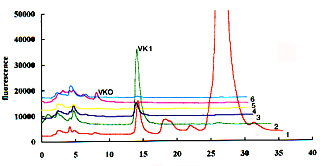
Detection limits are about 5-10 pg per injection but they depend on the sensitivity of the fluorescence detector.
Comments
A modified sensitive HPLC method, based on that described above, was developed for the determination of vitamin K1 in small sample (0.1-.25 ml) of plasma and serum (Wang LY et al., Clin Chim Acta 2004, 347, 199). A method for vitamin K derivatives (menaquinones) including the use of synthetic internal standards with various alky side-chain to increase the accuracy was also developed (Kamao M et al., J Chromatogr B 2005, 816, 41). A very sensitive procedure for quantification of vitamin K1 in human plasma by HPLC and a post-column on-line zinc metal reactor and a fluorometric detector has been described (Paroni R et al., J Chromatogr B 2009, 877, 351).
Preparation of vitamin K1 oxide: 100 µg of vitamin K1 are dissolved in 1 ml of ethanol and after addition of some mg of sodium carbonate and 3 µl oxygen peroxide (10%, w/w), the mixture is warmed for 30 min at 70°C with occasional agitation. After cooling, 1 ml water and 2 ml diethyl ether are added. After 5 min vortexing followed by a brief centrifugation, the upper phase is collected and evaporated, the dry residue being dissolved in a known volume of hexane or methanol.
Preparation of 2′,3′-dihydrovitamin K1: 10 mg of VK1 are dissolved in 150 ml of ethanol and warmed at 60°C. The mixture is flushed with nitrogen 2-3 min and 100 mg of catalyst (10% palladium on carbon) are added. Hydrogen is bubbled for 1 h. After cooling, 200 ml hexane are added, followed by 75 ml water, the mixture being agitated for 5 min. The hexane layer is collected and shaken with a solution of cerium(IV) sulphate to re-oxidize the hydroquinone formed. An aliquot of the hexane solution is chromatographed on a silica column (25 x 0.4 cm) with hexane/diisopropyl ether (99.5/0.5, v/v) as mobile phase. The fraction containing the VK1 derivative, detected at 250 nm), is concentrated by evaporation, solutions are kept in the cold (Langenberg JP et al., J Chromatogr 1984, 289, 377).
Determination of vitamin K1 isomers
As some cis-isomers may be formed during vitamin K synthesis or by photoisomerization, the individual measurement of the biologically active trans- and inactive cis-vitamin K1 should be estimated.
The use of the triacontyl-bonded C30 phase (polymeric YMC coumn) allowed a good separation of these isomers and furthermore allowed the estimation of contaminating menaquinone from bacterial origin (Woolard DC et al., J AOAC Int 2002, 85, 682). After lipase digestion to remove lipids, a reliable protocol was established for natural and processed foods. Thus, it was estimated that the cis-vitamin K1 isomer contributes up to about 15% of total phylloquinone in certain foodstuffs.
DISPERSIVE LIQUID-LIQUID MICROEXTRACTION
Lire la suiteDevenez membre et participez au développement de la Lipidomique au XXIème siècle.
S'inscrire