VITAMIN E ANALYSIS
The vitamin E complex is easily quantified provided some precautions are observed during the extraction of the biological tissues;
Extraction:
A- Extraction from fatty materials (liver, adipose tissues, oil, food…)
When samples are too fatty, a saponification step is needed to remove the excess of fatty acids before the HPLC analysis.
Saponification involves the heating of the sample in a strong alkaline medium, thus reducing the amount of organic compounds extractabled from the sample. Liberated fatty acids and alcohol will remain in the aqueous phase.
Up to 2 g of sample is saponified with 2 ml of KOH (60%, w/v), 2 ml ethanol, and 5 ml of ethanolic pyrogallol (6%, w/v). After a digestion time of 45 min at 70°C, the tubes are cooled and 15 ml of physiological saline are added. The mixture is extracted with 15 ml of hexane/ethyl acetate (9/1, v/v). The upper layer is evaporated and the dry residue is dissolved in a known volume of hexane/isopropanol (99/1, v/v).
Solid phase extraction (SPE) may be optimized for the extraction of tocopherols from vegetal oil before HPLC analysis (Grigoriadou D et al., Food Chem 2007, 105, 675). The study was shown to be useful in the official control of olive oil.
B- Extraction from cellular membranes, cellular suspensions, blood…
When samples contain only few amounts of triacylglycerols, an extraction with apolar solvents is suitable and the saponification step is not necessary.
Several methods are reliable.
When only vitamin E compounds are to be studied (tocopherols, tocotrienols) the rapid extraction process given for vitamin A is convenient. Vitamin A, carotenoids, and all apolar lipids may be also determined in this type of extract.
When the sample is liquid (plasma, cell suspension), the method is slightly modified in adding a different ethanol-SDS solution to the sample. At 1 ml of sample are added 1 ml of 2.5% sodium dodecyl sulfate in water and 2 ml ethanol. After vortexing 15-20 min in amber tubes, lipids are extracted after the addition of 3 ml hexane and vortexing 5 min. After a brief centrifugation, the upper hexane phase is evaporated and the residue is stored in a small amount of methanol before HPLC analysis. If the final solution is not limpid (because of triacylglycerols), the dry extract is re-dissolved in some µl of the mixture chloroform/methanol (1/3).
An optimized procedure for the extraction of tocopherols and tocotrienols from hazelnuts has been described (Amaral JS et al., Anal Sci 2005, 21, 1545). The procedure is based on a simple solid-liquid extraction using ethanol and hexane. A comparison of three method of extraction of vitamin E isomers from seeds and nuts followed by HPLC and coulometric detection may be found by Delgado-Zamarreno MM et al. (J Chromatogr A 2001, 935, 77). A large review of the different extraction procedures of fat-soluble vitamins including vitamin E from human fluids, foods and pharmaceutical preparations may be found by Luque-Garcia JL et al. (J Chromatogr A 2001, 935, 3).
When a joint analysis of vitamin E, cholesterol and polar lipids (phospholipids) is needed, all lipids are extracted according to the Folch’s procedure or, if necessary, to another procedure adapted to liquid samples.
For plasma or cell suspensions, we have described a reliable procedure which permits quantitative analysis of vitamin E, cholesterol and phospholipid fatty acids in a tiny biological sample (Leray C et al., J Chromatogr 1997,696, 33-42).
An alternative extraction procedure has been described which is simple and has a good reproducibility and recovery for tocopherols as well as retinol in plasma or serum (Taibi G et al., J chromatogr B 2002, 780, 261). A precise and sensitive procedure for simultaneous determination of all-trans-retinol and alpha-tocopherol in human serum following a two step extraction procedure has been proposed (Khan MI et al., Chromatographia 2010, 71, 577). A solid-phase extraction of vitamin E as well as Vitamin A in animal feeds was shown to give rapidly reproducible and precise results (Fedder R et al., J AOCS Int 2005, 88, 1579).
A reliable method has been developed for fish muscle and can be applied to similar biological marerials (Araujo M, et al., Eur J Lipid Sci Technol 2015, 117, 1398). Muscle samples are homogenized in a grinder, sample aliquots of approximately 1 g are
extracted with 5 mL of ethanol and 10 mL of n-hexane (0.01% of BHT). After vortexing and constant agitation (30 min), 5 mL of 1% NaCl solution are added. The mixture is centrifuged and the residue is re-extracted twice with 10 mL of n-hexane. Organic layers are pooled and Na2SO4 is added. After a new centrifugation, the supernatant is collected and the solvent dried under nitrogen stream. The residue is re-dissolved in 500mL of n-hexane and filtered before injection into the HPLC system.
Extraction: The cell suspension is centrifuged and the cell pellet is resuspended in 0.8 ml of 25 mM EDTA pH 7 (to prevent phospholipid degradation). Total lipids are extracted by vortexing 20 min after addition of 1 ml chloroform, 1 ml methanol and 50 µl of 15 mM BHT, and again 5 min after further additions of 1 ml chloroform and 1 ml NaCl to obtain two phases. After centrifugation, the lower phase is washed if necessary with 2 ml of 1M NaCl and evaporated under nitrogen.
Purification: Tocopherols, cholesterol and individual phospholipids are separated by one-dimensional TLC on Whatman LK5 silica gel plates. Plates were previously washed by migration up to the top in chloroform/methanol (1/1) in a special clean tank, dried and largely wetted with a solution of 2.5% boric acid in ethanol (except the concentration zone). After draining and dessication at 100°C (15 min), samples are applied as streaks in the concentration zone. The solvent system used is the same we have described for phospholipids but with addition of 10 mg/ml ascorbic acid (previously dissolved in water) and BHT (0.15 mg/ml) to protect tocopherols and unsaturated fatty acids against oxidation. The plates are developed up to 1 cm below the top in a dark chamber (the glass tank is under a plastic box). After drying, lipid spots are located under a UV lamp after a primulin spray (1 mg of primuline powder in 100 ml of a mixture acetone/water, 80/20). Commercial standard are used to locate the lipids of interest. Tocopherols and cholesterol have a quite similar migration and are found near the solvent front (average RF: 0.87).
The spot is scraped off and tocopherols eluted by vortexing some minutes with 3 ml of a mixture hexane/ter-butyl ether (92/8) or with pure ter-butyl ether if cholesterol must also be determined. The solvent phases are evaporated under nitrogen and the residue, dissolved in methanol, is stored in amber glass ware at -20°C.
HPLC: tocopherols (and cholesterol) are separated on a column packed with 5 µm LiChrosorb RP18 (Merck) (120×4.6 mm), with methanol/water (98/2) as the mobile phase, flow-rate: 1.5 ml/min.
Detection: tocopherols can be detected by light-scattering, spectrophotometry, fluorimetry and electrochemistry (cholesterol can be detected by light scattering and spectrophotometry). Tocopherol acetate (commonly used in food, dietetic and pharmaceutical preparations) is not fluorescent and may be detected by UV photometry or by light-scattering.
As response factors are different for the different tocopherols with any detector system, standard curves must be determined for each component.
With UV detection (292 nm for free tocopherols and 284 nm for tocopherol acetate), the minimum detection limit is about 15-30 ng (35-70 pmol) per injection. With fluorescence detection (excitation at 292 nm, emission at 327 nm) the minimum detection limit is about 0.5 ng (2-3 nmol) according to the instrument used. With the evaporative light-scattering detector, responses are linear in the range 0.1-0.5 µg and the minimum detection limits are about 10-20 ng per injection. These observations were made with a DDL21 detector (Eurosep Instruments) optimized with an air pressure set at 1 bar, an evaporation temperature at 60°C and a high voltage at 700 V. Other instruments should be optimized specifically.
d-tocopherol, rarely present in animal tissues, and tocopherol acetate are often used as internal standards to correct from losses during extraction and washings.
A typical chromatogram of a mixture of tocopherols obtained with a light-scattering detector is given below.
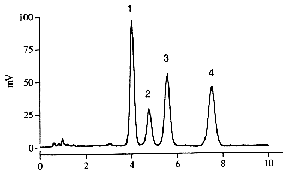
Peaks 1: δ-tocopherol, 2: γ-tocopherol, 3:α-tocopherol, 4: tocopherol acetate.
Time in minutes.
Several ways to perform sample pre-treatment and analysis of α-tocopherol may be found in the review by Ruperez FJ et al. (J Chromatogr A 2001, 935, 45). That work includes referenced tables providing in-depth summaries of methodology for the analysis of α-tocopherol and related compounds in foods, pharmaceuticals, plants, animal tissues and other matrices.
A comparison of chromatographic separation of tocopherols by reversed-phase, normal-phase, and gas chromatography may be found by Pyka A et al. (J Chromatogr A 2001, 935, 71).
Several methods have been developed for simultaneously determining α-tocopherol and cholesterol since they have both important dietary functions and furthermore share similar chromatographic properties. With the HPLC system described above, cholesterol eluted after about 9.5 min and is easily quantified with a light-scattering detector.
Another chromatographic system has been described involving a silica column eluted with an isocratic mobile phase made of isooctane/tetrahydrofuran (90/10, v/v) (Cayuela JM et al., J Agric Food Chem 2003, 51, 1120). A fluorescence detector was used to quantify tocopherols and a light-scattering detector to quantify cholesterol. Fluorescence was preferably used since, in the absence of previous purification of muscle extracts, this technique prevents interferences from unknown non-fluorescent substances eluting near α-tocopherol.
Very good and rapid separations of tocopherols were also obtained using nano-HPLC with a capillary column (100 mm ID) filled with 3 mm RP18 particles (Fanali S et al., J Pharm Biomed Anal 2004, 34, 331). Monolithic column were shown to improve the determination of vitamin E and A in human serum in shortening the time of analysis four times in comparison with using traditional particulate columns (Urbanek L et al., Anal Chim Acta 2006, 573, 267).
An example of simultaneous determination of a-tocopherol, b-carotene and retinol by HPLC with diode-array detection may be found in the work of Gimeno E et al. (J Chromatogr B 2001, 758, 315) as well as in the work of Casal S et al. (J Chromatogr B 2001, 763, 1).
The precise analysis of the carboxyethyl-hydroxychroman metabolites of α– and γ-tocopherol found in plasma or urine is possible when using a combination of gas chromatography with mass spectrometry (Galli F et al., Free Rad Biol Med 2002, 32, 333).
Analysis of tocotrienols and tocopherols
Several chromatographic HPLC methods were published and analyses involved either normal or reversed phase columns. Normal phase columns are more efficient to resolve all vitamin E vitamers (tocols) as they are found in foodstuffs.
One of the most reliable method for the simultaneous determination of tocopherols and tocotrienols is given below (Panfili G et al., J Agric Food Chem 2003, 51, 3940).
The vitamin E complex is extracted as described before for fatty materials with a hot saponification to provide the highest vitamin E recovery.
The chromatographic separation is achieved with a normal phase (Kromasil Phenomenex Si column) eluted with hexane/ethyl acetate/acetic acid (97.3/1.8/0.9, v/v) at a flow rate of 1.6 ml/min. Fluorometric detection is performed at an excitation wavelength of 290 nm and an emission wavelength of 330 nm.Tocopherols may be obtained from Merck and totrienols may be purified from barley extracts. The concentrations of the purified standards are calculated using the extinction coefficients given in this site.
A chromatogram of tocopherols and tocotrienols found in a barley sample is given below.
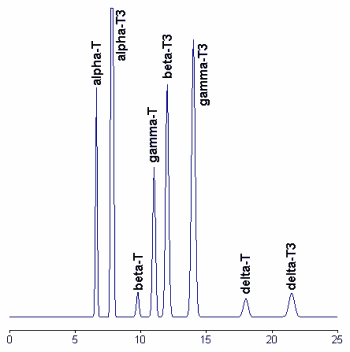
To improve the stability and the reproducibility of the silica column used, the column was flushed every eight injections with hexane/isopropanol (90/10, v/v). Tocotrienols standards are available from commercial sources but give double peaks due to the formation of racemic mixtures during synthesis. Thus, the preparation of pure vitamers from natural sources (barley, indian corn, and wheat) allows the formation of symmetrical peaks.
A base-line separation of all vitamers of tocopherol and tocotrienol has been obtained by capillary electrochromatography with an octylsilica column and acetonitrile/methanol as a mobile phase (Abidi SL et al., J Chromatogr A, 2001, 913, 379).
The determination of several geometrical isomers of tocotrienols was described after derivatization into methyl ether and chromatography on a chiral phase. Thus, a complete resolution of the eight isomers of α-tocotrienols was easily achieved in a 30 min run (Drotleff AM et al., J Chromatogr A 2001, 909, 215).
An efficient method to analyze tocopherols and tocotrienols, including tocomonoenol, was described using a C30 silica stationary phase and a fluorescence detector (Ng MH et al., Lipids 2004, 39, 1031).
Analysis of tocopherolquinone and epoxytocopherolquinones
These compounds are analyzed simultaneously with tocopherols or tocotrienols using an isocratic HPLC procedure combined with post-column zinc reduction and a single electrode coulometric detection (Leray C et al., J Lipid Res 1998, 39, 2099).
If pure compounds or clean extracts are analyzed, the monitoring of the elution may be made at 262 nm for tocopherolquinone and 270 nm for epoxydes. The column and eluant are similar to those used with UV or fluorescent detection of tocopherols but 5 mM ZnCl2, 2.5 mM Na acetate and 2.5 mM acetic acid are added to the mobile phase. A solid-phase post-column reactor similar to that used for the HPLC separation of vitamin K is used to reduce the quinone compounds before their coulometric detection in the oxidative mode. One electrode was used and set at a potential of + 600 mV. As for vitamin K, the mobile phase must be continuously deaerated by nitrogen gas bubbling one hour before flowing and during the run.
Below, a chromatogram of products of α-tocopherol oxidation is shown:
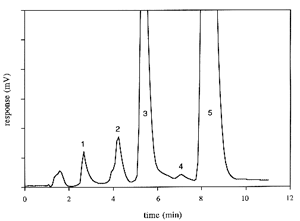
Peaks 1: tocopherolhydroquinone, 2: tocopherol quinone epoxyde, 3: tocopherolquinone, 4: γ-tocopherol and 5: α-tocopherol.
A similar procedure was used not only for vitamin E and its direct oxidation products but also for the addition products of α-tocopherol with peroxyl radicals derived from cholesteryl ester hydroperoxides and phosphatidylcholine hydroperoxides in plasma (Yamauchi R et al., Lipids 2002, 37, 515).
The determination of α-tocopherol and tocopherolquinone may also be effected by HPLC with electrochemical detection (Kanazawa H et al., Chem Pharm Bull 2000, 48, 1462). A direct and efficient procedure of tocopherol analysis in vegetable oils was described using differential pulse voltammetry (Diaz TG et al., Anal Chim Acta 2004, 511, 231).
The fluorescence detection of tocopherolquinone following derivatization by a photoreactor (excitation wavelength : 254 nm) has been shown to be an efficient alternative to the derivatization with a zinc reduction column (Pollok D et al., J Chromatogr A 2004, 1056, 257).
Preparation of tocopherolquinone
This compound can be prepared easily by Fe+++ oxidation in methanol solution (Schudel P et al Helv Chim Acta 1963, 46, 333).
To a solution of tocopherol in diethyl ether (1 g in 10 ml), add 2.5 ml of a solution made by dissolving FeCl3 (1.2 g in 15 ml methanol/water, 50/50, v/v). After 30 min agitation, the mixture is centrifuged and the lower phase is removed. Repeat this process 4 times and wash extensively the ether phase with water (8-10 times).
Evaporate the ether phase and dissolve in ethanol or ether.
Determine the exact concentration of a solution in measuring the absorbance at 260 nm of a mother solution (about 1 mg/ml ethanol) diluted 100 times. The molar extinction coefficient is taken as 19500 (260 nm).
The purity of the product is verified by HPLC as for tocopherols by injecting 10 µl of a tocopherolquinone solution (1 mg/ml ethanol) and recording the absorbance at 260 nm.
Preparation of tocopherolquinone epoxyde
(Liebler C et al Biochemistry 1992, 31, 8278)
Analysis of 5-nitro-γ-tocopherol
Extraction : the same procedures as for tocopherols
HPLC: nitro-g-tocopherol were separated from tocopherols on a deactivated octadecyl silane column (15 x 0.46 cm Supelcosil LC-18-DB, 3 mm particle size) eluted isocratically with 95/5 (v/v) methanol/0.5 M lithium acetate (pH 4.75) at a flow rate of 1.3 ml/min.
A coulometric detection was performed quantitatively at +500 mV (downstream electrode) after elimination of interfering substances at +300 mV (upstream electrode).
Below, a chromatogram of products of 5-nitro-γ-tocopherol (peak 3) and the two main tocopherols (peak 1 : γ-tocopherol, peak 2 : α-tocopherol.
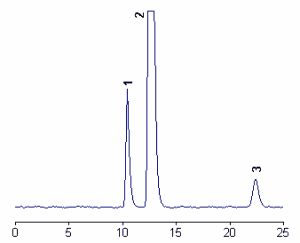
The detection of nitro-γ-tocopherol was linear over more than four orders of magnitudes from 0.1 pmol to 0.3 nmol, the detection limit being about 10 fmol (Christen S et al., J Lipid Res 2002, 43, 1978).
The isolation and quantification of 5-nitro-γ-tocopherol and the other reaction products of α-tocopherol with nitric oxide was done by HPLC and UV-visible detection but the detection by atmospheric pressure chemical ionization was shown to be more selective for these compounds (Nagata Y et al., J Chromatogr A 2004, 1036, 177).
Preparation of of 5-nitro-γ-tocopherol
This compound was synthesized by the nitrous acid method.
An ethanolic solution of γ-tocopherol (1 mg/ml) was acidified with 0.04 vol glacial acetic acid and nitration induced by the addition of 0.6 vol of a 2% sodium nitrite solution. After 2 min, the reaction was stopped with 0.4 vol of 20% potassium hydroxide. Two volumes of water were added and the crude product was extracted onto hexane and was purified by HPLC using 100% methanol as the eluent. The peak containing the pure product was collected after detection at 410 nm (extinction coefficient : 1,976 M-1 cm-1).
DISPERSIVE LIQUID-LIQUID MICROEXTRACTION
Lire la suiteDevenez membre et participez au développement de la Lipidomique au XXIème siècle.
S'inscrire